Improvement of Post-Exposure Delay Stability in Alicyclic ArF Excimer Photoresists
Introduction
Microlithography at 193 nm is one approach to extending optical lithography to sub 0.15 mm design rules. Photoresist materials for 193 nm lithography must fulfill the usual resist requirements such as high transparency at exposure wavelength and good resistance to dry etching processes. The relationship between the etch resistance of a polymer and the ratio of elements in its monomers is empirically described by the Ohnishi parameter[1].
Conventional resist materials for i-line and 248 nm lithography incorporate aromatic groups, which have relatively low Ohnishi parameters and provide good dry etch resistance. Unfortunately, the optical density of these materials is too high to be suitable for use at 193 nm. Therefore, the design of suitable photoresist materials for 193 nm lithography has been a significant challenge. Polymers incorporating alicyclic norbornene derivatives have shown great promise as candidates for 193 nm microlithographic application[2-14]. The alicyclic system has good transparency at 193 nm and useful etch resistance. In 1993, our group began studies on norbornene-derived amorphous polyolefins for use in 193 nm photoresist formulations. The first public description of this work was in 1996[3]. Since that time, a variety of photoresist formulations based on substituted alicyclic polymers have been reported. Resists based on polymers prepared by free-radical polymerization[3-11], cationic addition polymerization[3,6,12,13], and ring opening metathesis polymerization (ROMP)[6,12,14] have all been presented by our group and by others.
One of the more promising materials that has emerged from our work is the alternating copolymer, poly(DBNC-alt-MA), of t-butyl tetracyclo[4.4.0.12,5.17,10]dodec-3-ene-8-carboxylate (DNBC) and maleic anhydride (MA). This polymer can be easily prepared by free radical polymerization, Figure 1.
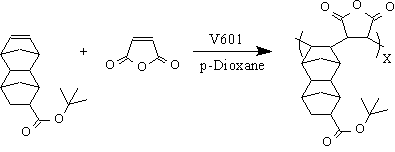
Figure 1: Synthesis of alternating copolymer, DNBC-alt-MA
Photoresist formulations based on this polymer have demonstrated good lithographic performance and acceptable dry etch resistance. Previous reports have shown how the performance of this material can be improved in several areas[8], but post-exposure delay stability has continued to be a problem.
These photoresists show two different modes of failure as a result of post-exposure delay. First, we observe an increase in linewidth with post-exposure delay and the formation of "t-topping" and skin. We believe that diffusion of the photogenerated acid in the bulk photoresist during the delay period leads to this variation of the final developed linewidth, and that contamination by airborne bases results in decreased efficiency of the photogenerated acid. However, this only affects a thin surface layer and therefore results in "t-topping" and skin. In order to improve the post-exposure delay stability of these resists, new polymers have been designed which address both of these manifestations of post-exposure delay.
A relatively large amount of free volume is trapped within a photoresist film during the spin-coating process as solvent molecules quickly evaporate. Trapped free volume allows increased diffusion of small molecules and leads to decreased post-exposure delay stability. Annealing a photoresist by heating during the post-application bake step to near its glass-transition temperature (Tg) allows the polymer molecules to relax to a more thermodynamically stable state, removing much of the trapped free volume. However, poly(DBNC-alt-MA) cannot be annealed without decomposition because the Tg exceeds the thermal stability of the t-butyl ester protecting groups. We therefore designed a variety of analogues in an attempt to adjust the Tg. Copolymers of the new 24O series shown in Figure 2 incorporate spacer groups between the acid-labile protecting group and dinorbornene units that lower their Tg allowing annealing of the resist film during the post-application bake step.
Our approach to addressing the "t-top" formation is exactly that which has proven so successful in the design of 248 nm photoresists. We simply increased the background dissolution rate by introducing an appropriate level of unblocked carboxylic acid comonomers.
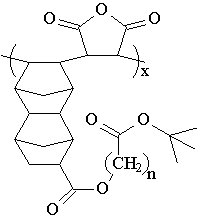
Figure 2: New annealable 24O series copolymer design
Experimental
A series of tert-butyl esters of w-bromo carboxylic acids, with alkyl chains of various lengths, were obtained according to the synthetic scheme shown in Figure 3[15].
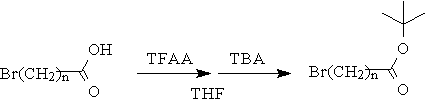
Figure 3: Synthesis of tert-butyl w-bromo- alkylcarboxylates
w-Bromoalkyl carboxylic acids, trifluoroacetic anhydride (TFAA), and tert-butyl alcohol (TBA, 2-methyl-2-propanol HPLC grade) were purchased from Aldrich Co. and used without further purification. A solution of w-bromoalkyl carboxylic acid in dry THF was cooled to 0 oC under a nitrogen atmosphere, and 1.5 equivalents of TFAA was added dropwise. The reaction mixture was then stirred for 1 hr maintaining a temperature below 15 oC. The reaction mixture was recooled to 0 oC, and 3 equivalents of TBA were added dropwise, followed by stirring overnight at room temperature under a nitrogen atmosphere. The reaction mixture was diluted with diethyl ether and washed with 3M NaOH, brine, and deionized water. The organic phase was then dried over MgSO4, and the solvents removed under reduced pressure to give the corresponding esters as clear or slightly yellow liquids which were used without further purification.
The dinorbornene monomers were obtained by esterification of tetracyclo[4.4.0.12,5.17,10]dodec-3-ene-8-carboxylic acid with the tert-butyl w-bromoalkyl carboxylates according to the scheme shown in Figure 4.
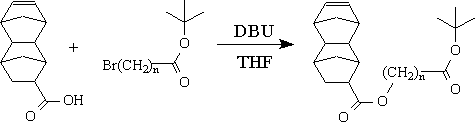
Figure 4: Synthesis of tetracyclo[4.4.0.12,5.17,10]dodec-3-ene-8-carboxylate derivatives
The reagent, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), was purchased from Aldrich Co. and tetracyclo[4.4.0.12,5.17,10]dodec-3-ene-8-carboxylic acid (DNCA) was obtained by the hydrolysis of methyl tetracyclo[4.4.0.12,5.17,10]dodec-3-ene-8-carboxylate, which was obtained from JSR Corporation. The esters were prepared by adding an excess of DBU dropwise to a solution of DNCA in dry THF under a nitrogen atmosphere at room temperature. The tert-butyl w-bromoalkyl carboxylate was added to the solution resulting in the formation of white crystals. The resulting mixture was stirred at room temperature under a nitrogen atmosphere overnight, then filtered and diluted with diethyl ether. The filtrate was washed with 3% HCl, 10% K2CO3, and deinonized water. After drying over MgSO4, the solvent was removed under reduced pressure to give esters as clear, viscous oils which were used without further purification.
The alternating polymers were prepared by radical polymerization. The dinorbornene monomer(s) and maleic anhydride were dissolved in dry p-dioxane with dimethyl 2,2�-azobisisobutyrate as a radical initiator (V601, Wako chemicals). Polymerization was carried out at 75 oC for 8 hrs under a nitrogen atmosphere. After the reaction was complete, an extra portion of p-dioxane was added to the polymer solution, and slowly added with stirring to an excess of cold hexane/IPA (50/50 by weight). The resulting precipitate was isolated by filtration, rinsed with the cold mixed solvent, air-dried, precipitated again under the same conditions, and vacuum dried at 50 oC overnight. The overall conversion ranged from 34-65%.
Triphenylsulfonium perfluorobutanesulfonate was prepared by a metathesis. Equimolar amounts of sodium perfluorobutanesulfonate and triphenylsulfonium bromide were partitioned between chloroform and deionized water by vigorously stirring the two-phase mixture for three hours. The organic phase was then seperated, dried over MgSO4, and the solvent was removed under reduced pressure to give triphenylsulfonium perfluorobutanesulfate as a oily residue. Exhaustive removal of the solvent gives the desired product as a white solid.
The monomers were characterized by NMR (General Electric, QE300) and IR spectra (Nicolet, Magna IR). Molecular weights were determined by gel permeation chromatography equipped with a refractive index detector (polystyrene standard, Viscotek). The glass transition temperature (Tg) of each alternating copolymer was measured by differential scanning calorimetery (Perkin Elmer, DSC7) (scan range=50-220 oC, scan rate=20 oC/min).
The photoresist solutions were coated over 52nm of DUV-30 ARC from Brewer Science. After spin-coating, the wafers were baked on a hotplate at 140 or 150 oC for 90 seconds. The resist films were exposed using an ISI 193nm Microstepper (0.6 NA, 0.7 s), and then baked at 140 or 150 oC for 90 sec. Development was performed using 2.38% tetramethylammonium hydroxide (TMAH) aqueous solution (Shipley CD-26) for 60 seconds in puddle mode.
Blanket etch rates were determined by measuring thickness loss observed during a fixed etch time in a LAM TCP 9400 prototype etcher with 300W chuck power and a gas mixture consisting of 150 sccm of HBr and 50 sccm of Cl2. The etch time was held constant because we have shown[7,8] that etch rates for polymers of this type are subject to large errors when determined by a single thickness measurement.
Results and Discussion
The annealable copolymers, represented by the structural formula shown in Figure 2, contain higher amounts of oxygen in their empirical formula. It was therefore important to determine the extent to which we had traded reduction of the Tg for dry etch resistance. Table 1 summarizes the characterization results of etching experiments on the 24O series copolymers. The etch rate of these new alternating copolymers are shown, normalized to that of the standard, poly(DBNC-alt-MA). We were pleased to see that all of the 24O series copolymers show an etch resistance very similar to that of poly(DBNC-alt-MA). There appears to be a slight dependence on the spacer chain length but the differences are so small that a statistical study would be required to clearly establish that relationship.
Table 1: Characterization results for 24O series polymers
|
Polymer |
Mw |
Pd |
Tg(oC) |
Etch rate |
|
24O-1 |
3790 |
1.52 |
190 |
1.01 |
|
24O-3 |
3340 |
1.47 |
125 |
1.06 |
|
24O-5 |
4130 |
1.47 |
110 |
1.10 |
|
24O-10 |
4380 |
1.49 |
100 |
1.09 |
|
DNBC-alt-MA |
4120 |
1.49 |
>250* |
1.00 |
24O-n, where n = spacer chain length
* Tg is higher than decomposition temperature of the t-butyl ester functionality
A plot of Tg vs. chain length data from Table 1 is shown in Figure 5.
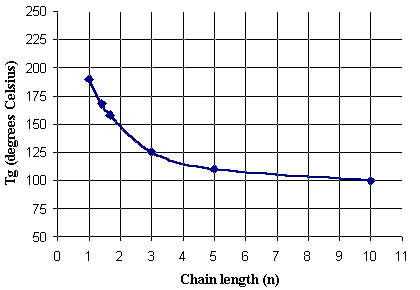
Figure 5: Tg versus spacer chain length
The Tg of the polymer decreases with increasing spacer chain length, but the influence of chain length plateaus when the chain length exceeds about n=5. Because of the bake conditions required to process resists of this type, we did not explore copolymers with a Tg below 125 �C, corresponding to a chain length of n = 3. Our goal was to produce copolymers with a Tg of approximately 140 oC. Unfortunately, that temperature corresponds to a chain length of approximately 2.5. We therefore blended these copolymers to precisely control the Tg. Terpolymers were also prepared from a mixture of appropriate 24O monomers with maleic anhydride. Both methods allowed us to effectively prepare polymers "non-integral" chain lengths.
Photoresist formulations for the initial imaging studies were prepared exactly like the standard formulation developed for poly(DBNC-alt-MA) photoresists[7,8] which consists of the polymer(s), triphenylsulfonium nonaflate as the photoacid generator, and 1-piperidineethanol as a base quencher. Both the post-application and post-exposure bakes were fixed at 140 oC for 90 seconds. Figure 6 shows images printed using different effective spacer chain lengths for the 24O series polymers. When n=3, an insoluble surface layer was observed that was not contamination related, but was rather the result of excessive hydophobicity.
Figure 6 also shows images obtained with blends of copolymers 24O-1 and 24O-3 that provided non-integral effective chain lengths. As the effective chain length is reduced, the problem of hydrophobicity is improved but the Tg begins to exceed the annealing range, so there is a narrow design window. The best images were obtained with a 1:2 blend ratio of 24O-3:24O-1 copolymers, giving an effective chain length of n=1.67 and a Tg of 156 �C. A terpolymer sample with the same effective chain length was synthesized by using a feed ratio of 2 : 1 : 3 of 24O-3 : 24O-1 : MA. The resulting terpolymer has the same Tg as the blend.

Figure 6: Effect of chain length, n, on the imaging quality of 24O series copolymers
On the basis of this data, a series of experiments were performed to optimize the resist formulations based on these new polymers. Several basic additives were auditioned as possible replacements for 1-piperidineethanol. Since this particular base was chosen for use with the standard poly(DBNC-alt-MA), it is possible that performance of the new 24O series polymers could be improved with a different base. Figure 7 shows the structure of the various bases tested. Although poly(DBNC-alt-MA) performs best with polar, cyclic amines such as 1-piperidineethanol, photoresists based on these new 24O-series copolymers perform best in conjunction with non-polar, trialkyl amines. For the remainder of imaging studies with these new polymers, 1-piperidineethanol was replaced with trioctylamine.
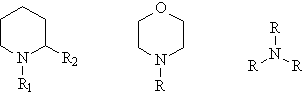
Figure 7: Structures of amines tested as possible improved basic additives
Having found a formulation based on annealable polymers that printed about as well as the standard formulation, we were now in a position to address the second mode of post-exposure delay failure. To that end, we explored the effect of adding small amounts of free acid to achieve a finite background dissolution rate. We knew from prior work that incorporating free acid into a polymer results in an increase in Tg. We therefore used the 24O-3 monomer alone to counteract the influence of the acid. Table 2 shows that incorporation of the 7-10% of free acid required to achieve a finite background dissolution rate gives a Tg of 155-158 �C, almost identical to that of the terpolymer or blend with n=1.67.
Table 2: Tg and thickness loss for acid containing terpolymers.
|
Acid feed
(%) |
Thickness loss (nm) |
Tg (oC) |
|
0 |
0.4 |
127 |
|
5 |
2.9 |
155 |
|
7.5 |
5.7 |
158 |
|
10 |
11.6 |
158 |
|
15 |
100.0 |
175 |
Thickness loss measured after 60 seconds develop time.
We examined the post-exposure delay stability of all of these materials. The measurements were made in comparison to a standard UT formulation based on poly(DBNC-alt-MA). Each wafer was coated, baked, and exposed without delay, followed by storage in open cassettes. The measured NH3 concentration in the facility was ~4ppb. Both the post-application and post-exposure bakes were fixed at 150 oC for 90 seconds, a temperature close to the Tg of the polymers with effective n=1.67. Figure 8 shows the results of these studies. Note that the standard formulation shows t-topping after only 10 minutes, and by 40 minutes, the linewidth has also increased dramatically. The annealable formulations, both the blend and the terpolymer, show little improvement in t-topping, but suffer minimal linewidth change even after 40 minutes of delay. As we had hoped, incorporation of a controlled amount of background dissolution removes the t-topping and gives excellent image quality with storage stability in excess of 40 minutes. Delay times as high as 100 minutes are necessary before "t-top" formation becomes evident with formulations that incorporate 10 % DNCA.
|
No Delay |
10 Minute
Delay |
20 Minute
Delay |
40 Minute
Delay |
100 Minute
Delay |
I |

|
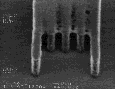
|

|

|
|
II |

|
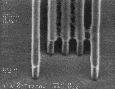
|

|

|
|
III |

|

|

|
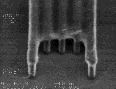
|
|
IV |

|

|
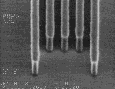
|
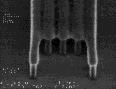
|
|
V |

|

|

|

|
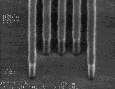
|
Figure 8: Post-exposure delay test results showing 160 nm images with a 1:1 pitch. (I)Standard formulation of poly(DBNC-alt-MA). (II)Blend polymer of 24O-1 and 24O-3 with effective n=1.67. (III)Terpolymer of 24O-1, 24O-3, and MA with effective n=1.67. (IV)Terpolymer of 24O-3, DNCA, and MA, acid feed concentration=7.5%. (V)Terpolymer of 24O-3, DNCA, and MA, acid feed concentration=10%.
Conclusions
It is not possible or appropriate to state that we have a full molecular-level understanding of the processes responsible for t-topping and linewidth change during post-exposure storage of chemically-amplified photoresists. However, assuming that the linewidth change is largely the result of acid diffusion and that t-top formation is largely the result of gettering of contaminants from the atmosphere has allowed us to rationally modify the design of our 193 nm photoresists to improve storage stability. Reducing the glass-transition temperature and adding a controlled amount of background dissolution have allowed us to extend the storage stability to over one hour in a typical cleanroom environment.
Acknowledgements
We are indebted to both Sematech (Contract #36041900) and SRC (Contract #96-LP-409) for their generous support of this project. We also would like to thank JSR Corporation for their support of Mikio Yamachika and for generous supply of monomer precursors.
References
- Goken, H.; Esho, S.; Ohnishi Y., J. Electrochem. Soc. 1983, 130, 423.
- Allen, R.D.; Wan, I.Y.; Wallraff, G.M.; DiPetro, R.A.; Hofer, D.; Kunz, R.R., J. Photopolymer Sci. Technol. 1983, 8, 623.
- Allen, R.; Sooriyakumaran, R.; Optiz, J.; Wallraff, G.; Kunz, R.; Jayaraman, S.; Schick, R.; Goodall, B.; Okoroanyanwu, U; Willson, C.G., Proc. SPIE 1996, 2724, 334.
- Okoroanyanwu, U.; Shimokawa, T.; Medeiros, D.; Willson, C.G.; Byers, J.; Niu, Q.J.; Frechet, J.M.J.; Allen, R.D., Proc. SPIE 1997, 3049, 272.
- Okoroanyanwu, U.; Byers, J.D.; Shimokawa, T.; Willson, C.G., Chem. Mater. 1998, 10, 3328.
- Okoroanyanwu, U. Ph.D. Thesis, The University of Texas at Austin, 1997.
- Patterson, K.; Okoroanyanwu, U.; Shimokawa, T.; Cho, S.; Byers, J.; Willson, C.G., Proc. SPIE 1998, 3333, 425.
- Byers, J.; Patterson, K.; Cho, S.; McCallum, M.; Willson, C. G.; J. Photopolymer Sci. Technol. 1998, 11, 465.
- Cho, S.; Patterson, K.; Byers, J.; Yamachika, M.; Yamada, S.; Rager, T.; Tran, H.; Willson, C. G., Technical Proceedings, Techcon 98 (Semiconductor Research Corporation Annual Meeting), 1998.
- Nozaki, K.; Yano, E., J. Photopolymer Sci. Technol. 1997, 10, 545.
- Jung, J.C.; Bok, C.H.; Baik, K.H. Proc. SPIE 1998, 3333, 11.
- Houlihan, F.M.; Wallow, T.I.; Nalamasu, O.; Reichmanis, E., Macromolecules 1997, 30, 6517.
- Allen, R.D.; Wallow, T.I.; Opitz, J.; Dipietro, R.; Hofer, D.; Jayaraman, S.; Hullihan, K.; Rhodes, L.; Goodall, B.; Shick, R. Proc. SPIE 1998, 3333, 463.
- Suwa, M.; Kajita, T.; Iwasawa, H.; Yamamoto, M. Proc. SPIE 1998, 3333, 11.
- Bonar-Law, R.P.; Davis, A.P.; Sanders, K.M.; J.Chem. Soc. Perkin Trans. 1. 1990, 2245.